Abstract
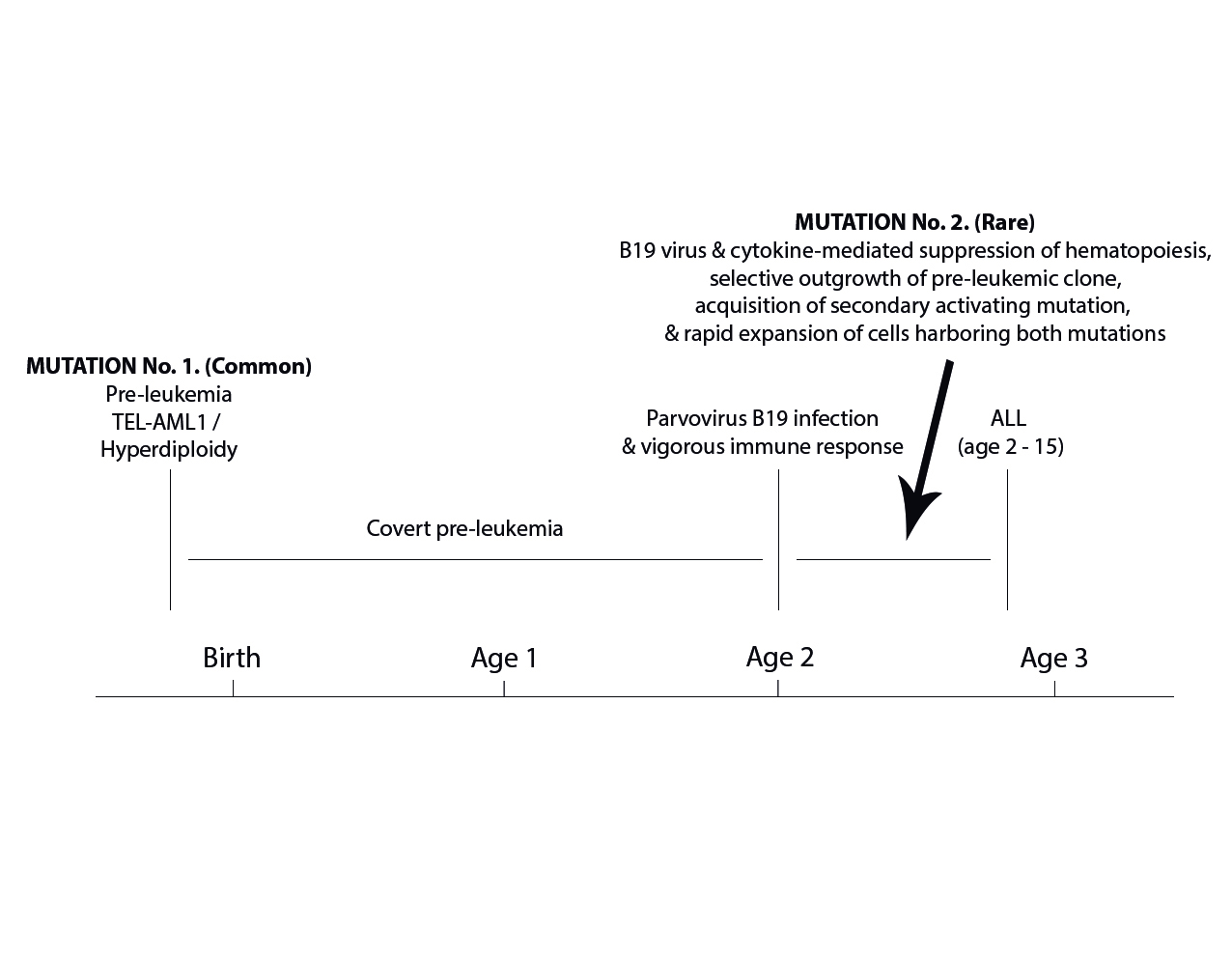
In this article, we review the evidence suggesting a possible role for B19 virus in the pathogenesis of a subset of cases of acute leukemia. Human parvovirus B19 infection may complicate the clinical course of patients with acute leukemia and may also precede the development of acute leukemia by up to 180?days. Parvovirus B19 targets erythroblasts in the bone marrow and may cause aplastic crisis in patients with shortened-red cell survival. Aplastic crisis represents a prodrome of acute lymphoblastic leukemia in 2% patients. There is a significant overlap between those HLA classes I and II alleles that are associated with a vigorous immune response and development of symptoms during B19 infection and those HLA alleles that predispose to development of acute leukemia. Acute symptomatic B19 infection is associated with low circulating IL-10 consistent with a vigorous immune response; deficient IL-10 production at birth was recently found to be associated with subsequent development of acute leukemia. Anti-B19 IgG has been associated with a particular profile of methylation of human cancer genes in patients with acute leukemia, suggesting an additional hit and run mechanism. The proposed role for parvovirus B19 in the pathogenesis of acute leukemia fits well with the delayed infection hypothesis and with the two-step mutation model, which describes carriage of the first mutation prior to birth, followed by suppression of hematopoiesis, which allows rapid proliferation of cells harboring the first mutation, acquisition of a second activating mutation, and expansion of cells carrying both mutations, resulting in acute leukemia. Copyright © 2015 John Wiley & Sons, Ltd.